If you design, test, or certify medical electrical equipment, the jump from the 2nd to the 3rd Edition of IEC 60601-1 isn’t just a paperwork update. It changed how you have to think about safety from the ground up.
The 2nd Edition treated safety like a checklist. You ran the tests, you passed or failed, and that was mostly the end of the conversation. The 3rd Edition asks a harder question: have you actually thought through everything that could go wrong with your device, and can you prove it?
That shift in mindset is the heart of this article. I’ll walk you through what changed, why it matters, and what it means for your project. This guide is written for design engineers, QA and RA teams, compliance managers, and product leads who need a clear, practical picture without wading through the full standard.
Here’s the short version: the 3rd Edition changes how you prove safety, not only what you test. Keep that in mind as we go.
What IEC 60601-1 Covers
IEC 60601-1 is the general safety standard for medical electrical equipment. Its job is to make sure these devices deliver two things: basic safety and essential performance. Basic safety means the device won’t shock, burn, or physically harm anyone under normal use or a single fault. Essential performance means the device continues to do the job it needs to do without creating an unacceptable risk.
The scope is broad. It covers everything from patient monitors and infusion pumps to surgical tools and lab-adjacent equipment that touches patient care. If a device plugs in or runs on stored power and interacts with a patient or operator, it’s usually in scope.
The standard doesn’t work alone, either. It sits at the top of a larger family. IEC 60601-1 is the general standard. Below it sit collateral standards that apply across many device types, and particular standards that target specific devices. Understanding that structure makes the edition changes much easier to follow, because the 3rd Edition pulls all three layers closer together.
Why the 3rd Edition Was a Major Shift
The 2nd Edition was prescriptive. It told you exactly what to test and what limits to meet, mostly around fixed hardware requirements. You measured leakage current, checked dielectric strength, confirmed the enclosure held up, and moved on. Pass or fail.
That worked fine when devices were simpler. But modern equipment is full of software, complex electronics, and connected systems. A pure pass/fail hardware test can’t catch every way a smart device might fail and hurt someone.
So the 3rd Edition built itself around risk. Instead of just checking boxes, you now have to identify what could go wrong, judge how serious it is, and show how your design controls it. Safety became something you manage across the entire design lifecycle, not a single gate you clear at the end.
If you remember one thing from this section, make it this: safety is now a continuous process, not a one-time test result.
Risk Management Became Central
The 3rd Edition ties directly to ISO 14971, the standard for risk management of medical devices. You can’t separate the two anymore. To meet IEC 60601-1, you need a formal risk management process running underneath your design work.
That means identifying hazards, estimating the risk each one carries, and documenting exactly how your design reduces that risk to an acceptable level. Every safety claim you make should trace back to that analysis. It’s not enough to say a feature is safe. You have to show the reasoning and the evidence.
This changes how engineering teams work. Risk thinking has to start early, not after the prototype is built. The earlier you bake it in, the fewer expensive surprises you hit later.
Why Test Labs Now Review More Than Test Data
Here’s a practical change that catches many teams off guard. Under the 2nd Edition, a lab could issue a certificate largely based on test results. Numbers passed, certificate issued.
That’s no longer how it works. A compliance lab now reviews your risk analysis, too. They want to see your risk management file and how it links to your safety claims. Test data alone won’t get you a certificate.
For your timeline, this matters a lot. If you show up to the lab with great test results but a thin or messy risk file, you’ll hit delays. Get your documentation in order before you book lab time. A clean, well-organized risk file can be the difference between a smooth assessment and weeks of back-and-forth.
Essential Performance as a Core Requirement
The 2nd Edition focused on preventing physical harm: electric shock, fires, burns, and mechanical injuries. That’s still important. But the 3rd Edition added a second pillar called essential performance.
Essential performance is the set of functions your device must continue to perform to remain safe. Put another way, if a function fails or degrades, and that creates an unacceptable risk to the patient, that function is part of your essential performance.
Think of an infusion pump. Preventing electric shock is basic safety. Delivering the right dose at the right rate is essential performance. If the pump keeps running but delivers the wrong amount, nobody got shocked, but the patient is still in danger.
So you have to define what your device “must do” to remain safe, then prove those functions hold up. That includes testing them under stress conditions, not just on a calm bench on a good day. This often reshapes your whole test plan, because you’re now validating the function, not just measuring electrical limits.
MOPP and MOOP Replaced Older Protection Concepts
The 2nd Edition used a broad term, “user,” to describe anyone near the device. The 3rd Edition got more specific, and that specificity drives real design decisions. It splits protection into two categories based on who you’re protecting.
Means of Patient Protection (MOPP)
MOPP covers protection for the patient receiving treatment. Patients are often the most vulnerable people in the room. They may be sedated, unconscious, connected to the device through electrodes or fluids, or simply unable to react to a malfunction.
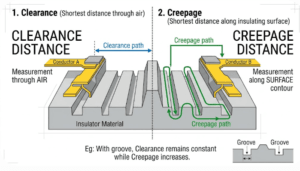
As a result, MOPP requirements are usually stricter. Isolation distances, insulation barriers, and leakage limits are tighter when a patient is the one being protected. The standard assumes the patient can’t protect themselves, so the device has to do more.
Means of Operator Protection (MOOP)
MOOP covers the people running the equipment: nurses, technicians, and clinicians. These users are generally healthy, alert, and able to step back if something goes wrong.
Because operators can react, MOOP requirements are often less demanding than MOPP. The split matters in design because it lets you apply the right level of protection to the right barrier. You don’t have to over-engineer every isolation point to patient-grade levels. You match the protection to who’s actually at risk, which can save cost and space without cutting safety.
Expanded Electrical, Mechanical, and System Requirements
The 3rd Edition didn’t just add risk thinking. It also tightened and broadened the technical requirements to match modern devices.
On the electrical side, the testing parameters for leakage current and dielectric strength were updated to reflect today’s components and circuit designs. Mechanical safety also received more attention, with more detailed checks for instability, movement of mobile equipment, and crushing hazards from moving parts or heavy assemblies.
The standard also pushes you to evaluate energy hazards more broadly in your risk file. That includes heat, radiation, and pressure systems, not just electricity. If your device stores or releases energy in any form, it needs to be on your radar.
Finally, the 3rd Edition clarified requirements for accessories and connected systems. Your device might be safe on its own, but the moment you connect it to a cart, a docking station, or another instrument, the whole setup must remain safe. You’re responsible for the system, not just the box.
Software and Programmable Electrical Medical Systems (PEMS)
Software earned its own dedicated attention in the 3rd Edition, and for good reason. So much of what makes modern devices safe lives in code, not just in hardware.
Clause 14 lays out requirements for programmable electrical medical systems. It covers the validation and verification steps you need to follow for software-driven functions. You can’t just trust that the code works. You have to show how you confirmed it.
That means documenting your software development process and conducting a risk analysis of software-specific failures, such as bugs, logic errors, or unexpected states. These problems don’t show up on a leakage current meter, so they need their own discipline.
These requirements line up closely with IEC 62304, the standard for medical device software lifecycle processes. If you’re already following IEC 62304, you’re well on your way. The two standards reinforce each other, and treating software reliability as part of overall compliance is now the expectation, not a bonus.
Collateral and Particular Standards in Practice
Compliance with the 3rd Edition is rarely about a single document. In practice, you’re usually meeting a package of standards across three layers.
The general standard is IEC 60601-1 itself. Collateral standards add requirements that apply across many device types, covering areas such as EMC and usability. Particular standards add requirements for specific kinds of devices. To get to market, you often need all three working together.
Key Collateral Standards to Know
A few collateral standards come up again and again:
- IEC 60601-1-2 covers electromagnetic compatibility (EMC). It sets immunity and emission limits so your device works safely around other electronics and doesn’t interfere with them.
- IEC 60601-1-6 covers usability engineering. It addresses use-related risks, making sure the interface design doesn’t lead to dangerous mistakes.
- IEC 60601-1-11 covers the home healthcare environment. If your device gets used outside a clinical setting, this one adds requirements for those less-controlled conditions.
Choosing the Right Standard
Specific standards are in the 60601-2-x series, and each targets a specific device type. There are standards for high-frequency surgical equipment, one for infusion pumps, one for patient monitors, and many more.
The job here is simple to state but easy to overlook: match your device to the correct particular standard. That standard will add tailored requirements and tests on top of the general and collateral standards. Skip it, and you’ll have a compliance gap that surfaces at the worst possible time.
Documentation and Technical File Requirements
The 3rd Edition raised the bar for documentation significantly. If you’re coming from the 2nd Edition, expect to produce a lot more paper or a lot more well-organized files.
The Risk Management File (RMF) is now the anchor of your technical file. It’s arguably the most important document you’ll create, because it links your test results to your safety claims and shows the reasoning behind both. Auditors and labs go straight to it.
You also need a traceability matrix. This document maps each requirement in the standard to a specific design feature or test that addresses it. It sounds tedious, but it’s incredibly useful. When an auditor asks how you meet a given clause, you point to the matrix and show the line. No scrambling.
Strong documentation does more than satisfy auditors. It makes your whole certification process smoother, faster, and far less stressful. The teams that treat documentation as a core deliverable, not an afterthought, are the ones that sail through assessments.
What the Change Means for Legacy and New Devices
If you have a device already on the market, you’re probably wondering whether you have to redo everything. The answer depends on the market and the situation.
In some cases, a legacy device certified to the 2nd Edition may still be accepted, at least for a transition period, in certain markets. But that window narrows over time, and most regulators have moved firmly to the 3rd Edition.
A common question is whether you need to retest a device when the physical components haven’t changed. Even then, you may need to revisit your risk management file and documentation to meet 3rd Edition expectations. The hardware might pass, but the paperwork standard is higher now.
Handling Mixed-Edition Realities
Here’s a wrinkle that trips up a lot of teams. The general standard and the particular standards don’t always move to a new edition at the same time. You can end up with IEC 60601-1 on the 3rd Edition, while a relevant particular standard still references an older edition.
Transition and harmonization guidance exists to help you manage these gaps. The key is not to assume everything is aligned. Check the editions of every standard that applies to your device.
You also have to confirm which editions your target markets actually recognize. The US, EU, and Canada don’t always accept the same versions on the same timeline. A device that’s fine in one region might experience a delay in another due to an edition mismatch. Verify before you commit.
How to Prepare for 3rd Edition Compliance
If you’re getting ready for the 3rd Edition, a little planning saves a lot of pain. Here’s where I’d start:
- Audit your risk management process first. Before you book any lab time, make sure your ISO 14971 process is solid, and your risk file is complete. This is the foundation everything else rests on.
- Align your teams early. Engineering, QA, and regulatory need to be on the same page from the start. Risk-based compliance fails when these groups work in silos.
- Confirm the standards that apply. Pin down the collateral and particular standards for your specific device, and check their editions.
- Check national deviations. Review the specific requirements for the US, EU, and Canada to avoid surprises that could delay your launch.
- Build for the future. The 4th Edition is already in development. A strong, risk-based framework now makes the next transition far easier.
Common FAQs About IEC 60601-1 3rd Edition
Is the 2nd Edition still valid for legacy devices in major markets?
In most major markets, the 2nd Edition has been phased out, though some legacy devices may still be accepted under specific transition arrangements. Don’t assume it’s still valid. Check the current rules for each market where you sell, because the acceptance windows have largely closed.
How does the 3rd Edition affect the FDA 510(k) submission process?
The FDA recognizes IEC 60601-1 3rd Edition, so a 510(k) submission generally needs to show compliance with it, including the risk management elements. That means your submission should include solid risk documentation, not just test reports. Expect reviewers to look for that risk-based evidence.
What’s the difference between Amendment 1 and Amendment 2 of the 3rd Edition?
Amendment 1 refined and clarified many requirements in the 3rd Edition, including those related to risk management and essential performance. Amendment 2 brought further updates and corrections. The practical takeaway: confirm which amendment level your target market requires, since that affects exactly what you need to meet.
Does a device need retesting if its physical components haven’t changed?
Possibly. Even with unchanged hardware, you may need to update your risk management file and documentation to satisfy 3rd Edition requirements. Some testing may not need to be repeated, but the documentation and risk analysis usually need to meet newer expectations.
Which IEC 60601-1-2 standards are mandatory for compliance?
It depends on your device and market, but IEC 60601-1-2 for EMC is almost always required. Others, such as IEC 60601-1-6 for usability or IEC 60601-1-11 for home use, apply depending on how and where the device is used. Identify the ones tied to your device early.
Conclusion
The move from the 2nd to the 3rd Edition reshaped how the industry proves medical device safety. The biggest changes all connect: a risk-based approach replaced the old checklist; essential performance joined basic safety; MOPP and MOOP brought clarity on who you’re protecting; software and PEMS earned real attention; and documentation expectations climbed sharply.
Every one of those changes points back to a single goal: protecting patients and operators. The standard became more demanding as devices grew more complex, and the old methods couldn’t keep up.
If you’re navigating this transition, plan early, document with discipline, and keep a clear view of which standards and editions apply to your device. And before you commit to a design or a market, confirm exactly which editions and amendments that market accepts. A few hours of upfront checking can save you months of delay down the line.